All in One View
Content from Data Tidiness
Last updated on 2023-06-14 | Edit this page
Estimated time: 30 minutes
Overview
Questions
- What metadata should I collect?
- How should I structure my sequencing data and metadata?
Objectives
- Think about and understand the types of metadata a sequencing experiment will generate.
- Understand the importance of metadata and potential metadata standards.
- Explore common formatting challenges in spreadsheet data.
Introduction
When we think about the data for a sequencing project, we often start by thinking about the sequencing data that we get back from the sequencing center. However, equally or more important is the data you’ve generated about the sequences before it ever goes to the sequencing center. This is the data about the data, often called the metadata. Without the information about what you sequenced, the sequence data itself is useless.
Discussion
With the person next to you, discuss:
What kinds of data and information have you generated before you sent your DNA/RNA off for sequencing?
Types of files and information you have generated:
- Spreadsheet or tabular data with the data from your experiment and whatever you were measuring for your study.
- Lab notebook notes about how you conducted those experiments.
- Spreadsheet or tabular data about the samples you sent off for sequencing. Sequencing centers often have a particular format they need with the name of the sample, DNA concentration and other information.
- Lab notebook notes about how you prepared the DNA/RNA for sequencing and what type of sequencing you’re doing, e.g. paired end Illumina HiSeq. There likely will be other ideas here too. Was this more information and data than you were expecting?
All of the data and information just discussed can be considered metadata, i.e. data about the data. We want to follow a few guidelines for metadata.
Notes
Notes about your experiment, including how you prepared your samples for sequencing, should be in your lab notebook, whether that’s a physical lab notebook or electronic lab notebook. For guidelines on good lab notebooks, see the Howard Hughes Medical Institute “Making the Right Moves: A Practical Guide to Scientifıc Management for Postdocs and New Faculty” section on Data Management and Laboratory Notebooks.
Ensure to include dates on your lab notebook pages, the samples themselves, and in any records about those samples. This will help you correctly associate samples other later. Using dates also helps create unique identifiers, because even if you process the same sample twice, you do not usually do it on the same day, or if you do, you’re aware of it and give them names like A and B.
Unique identifiers
Unique identifiers are a unique name for a sample or set of sequencing data. They are names for that data that only exist for that data. Having these unique names makes them much easier to track later.
Data about the experiment
Data about the experiment is usually collected in spreadsheets, like Excel.
What type of data to collect depends on your experiment and there are often guidelines from metadata standards.
Metadata standards
Many fields have particular ways that they structure their metadata so it’s consistent and can be used across the field.
The Digital Curation Center maintains a list of metadata standards and some that are particularly relevant for genomics data are available from the Genomics Standards Consortium.
If there are not metadata standards already, you can think about what the minimum amount of information is that someone would need to know about your data to be able to work with it, without talking to you.
Structuring data in spreadsheets
Regardless of the type of data you’re collecting, there are standard ways to enter that data into the spreadsheet to make it easier to analyze later. We often enter data in a way that makes it easy for us as humans to read and work with it, because we’re human! Computers need data structured in a way that they can use it. So to use this data in a computational workflow, we need to think like computers when we use spreadsheets.
The cardinal rules of using spreadsheet programs for data:
- Leave the raw data raw - do not change it!
- Put each observation or sample in its own row.
- Put all your variables in columns - the thing that vary between samples, like ‘strain’ or ‘DNA-concentration’.
- Have column names be explanatory, but without spaces. Use ‘-’, ‘_’ or camel case instead of a space. For instance ‘library-prep-method’ or ‘LibraryPrep’is better than ’library preparation method’ or ‘prep’, because computers interpret spaces in particular ways.
- Do not combine multiple pieces of information in one cell. Sometimes it just seems like one thing, but think if that’s the only way you’ll want to be able to use or sort that data. For example, instead of having a column with species and strain name (e.g. E. coli K12) you would have one column with the species name (E. coli) and another with the strain name (K12). Depending on the type of analysis you want to do, you may even separate the genus and species names into distinct columns.
- Export the cleaned data to a text-based format like CSV (comma-separated values) format. This ensures that anyone can use the data, and is required by most data repositories.
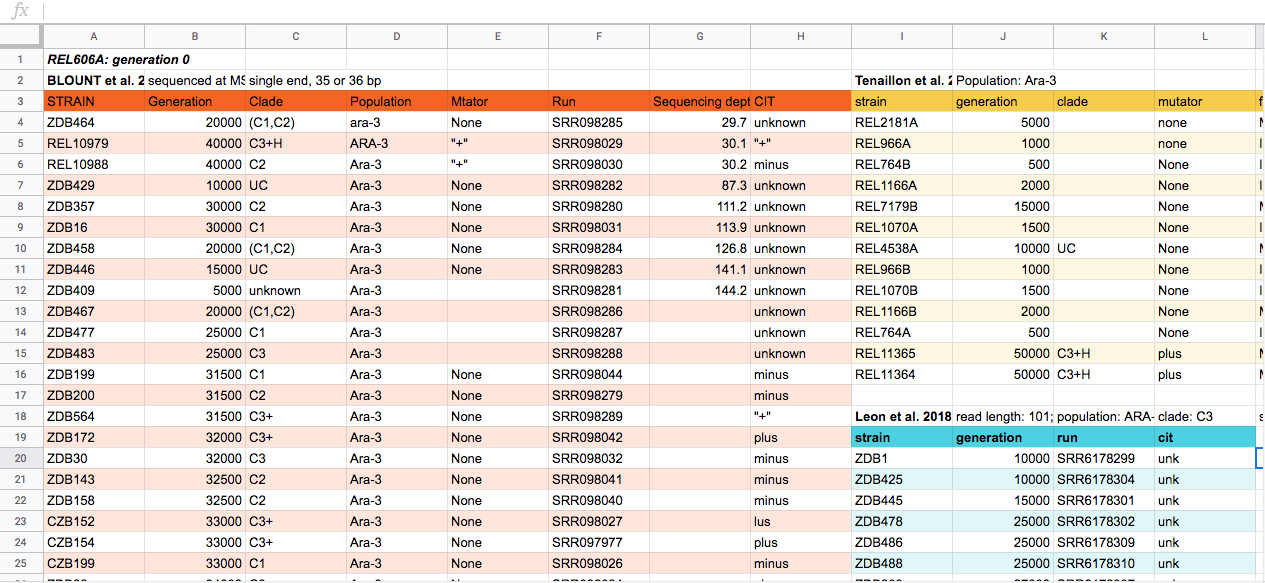
Discussion
This is some potential spreadsheet data generated about a sequencing experiment. With the person next to you, for about 2 minutes, discuss some of the problems with the spreadsheet data shown above. You can look at the image, or download the file to your computer via this link and open it in a spreadsheet reader like Excel.
A full set of types of issues with spreadsheet data is at the Data Carpentry Ecology spreadsheet lesson. Not all are present in this example. Discuss with the group what they found. Some problems include not all data sets having the same columns, datasets split into their own tables, color to encode information, different column names, spaces in some columns names. Here is a “clean” version of the same spreadsheet:
Cleaned spreadsheet Download the file using right-click (PC)/command-click (Mac).
Further notes on data tidiness
Organizing your data properly at this point of your experiment will help your analysis later. It will also prepare your data and notes for data deposition, which is often required by journals and funding agencies. If this is a collaborative project, as most projects are now, it’s also vital information for your collaborators. Well organized data is very useful for communication and efficiency.
Fear not! If you have already started your project and it’s not set up this way, there are still opportunities to make updates. One of the biggest challenges is tabular data that is not formatted so computers can use it, or has inconsistencies that make it hard to analyze.
More practice on how to structure data is outlined in our Data Carpentry Ecology spreadsheet lesson
Tools like OpenRefine can help you clean your data.
- Metadata is key for you and others to be able to work with your data.
- Tabular data needs to be structured to be able to work with it effectively.
Content from Planning for NGS Projects
Last updated on 2024-04-04 | Edit this page
Estimated time: 30 minutes
Overview
Questions
- How do I plan and organize a genome sequencing project?
- What information does a sequencing facility need?
- What are the guidelines for data storage?
Objectives
- Understand the data we send to and get back from a sequencing center.
- Make decisions about how (if) data will be stored, archived, shared, etc.
There are a variety of ways to work with a large sequencing dataset. You may be a novice who has not used bioinformatics tools beyond doing BLAST searches. You may have bioinformatics experience with other types of data and are working with high-throughput (NGS) sequence data for the first time. In the most important ways, the methods and approaches we need in bioinformatics are the same ones we need at the bench or in the field - planning, documenting, and organizing are the key to good reproducible science.
Discussion
Before we go any further, here are some important questions to consider. If you are learning at a workshop, please discuss these questions with your neighbor.
Working with sequence data
What challenges do you think you’ll face (or have already faced) in working with a large sequence dataset? What is your strategy for saving and sharing your sequence files? How can you be sure that your raw data have not been unintentionally corrupted? Where/how will you (did you) analyze your data - what software, what computer(s)?
Sending samples to the facility
The first step in sending your sample for sequencing will be to complete a form documenting the metadata for the facility. Take a look at the following example submission spreadsheet.
Download the file using right-click (PC)/command-click (Mac). This is a tab-delimited text file. Try opening it with Excel or another spreadsheet program.
Exercise
- What are some errors you can spot in the data? Typos, missing data, inconsistencies?
- What improvements could be made to the choices in naming?
- What are some errors in the spreadsheet that would be difficult to spot? Is there any way you can test this?
Errors:
- Sequential order of well_position changes
- Format of client_sample_id changes and cannot have spaces, slashes, non-standard ASCII characters
- Capitalization of the replicate column changes
- Volume and concentration column headers have unusual (not allowed) characters
- Volume, concentration, and RIN column decimal accuracy changes
- The prep_date and ship_date formats are different, and prep_date has multiple formats
- Are there others not mentioned?
Improvements in naming
- Shorten client_sample_id names, and maybe just call them “names”
- For example: “wt” for “wild-type”. Also, they are all “1hr”, so that is superfluous information
- The prep_date and ship_date might not be needed
- Use “microliters” for “Volume (µL)” etc.
Errors hard to spot:
- No space between “wild” and “type”, repeated barcode numbers, missing data, duplicate names
- Find by sorting, or counting
Retrieving sample sequencing data from the facility
When the data come back from the sequencing facility, you will receive some documentation (metadata) as well as the sequence files themselves. Download and examine the following example file - here provided as a text file and Excel file:
Exercise
- How are these samples organized?
- If you wanted to associate the sequence file names with their corresponding sample names from the submission sheet, could you do so? How?
- What do the _R1/_R2 extensions mean in the file names?
- What does the ‘.gz’ extension on the filenames indicate?
- What is the total file size - what challenges in downloading and sharing these data might exist?
- Samples are organized by sample_id
- To relate filenames use the sample_id, and do a VLOOKUP on submission sheet
- The _R1/_R2 extensions mean “read 1” and “read 2” of each sample. These typically refer to forward and reverse reads of the same DNA fragment from the sequencer, i.e. during paired-end sequencing.
- The ‘.gz’ extension means it is a compressed “gzip” type format to save disk space
- The size of all the files combined is 1113.60 Gb (over a terabyte!). To transfer files this large you should validate the file size following transfer. Absolute file integrity checks following transfers and methods for faster file transfers are possible but beyond the scope of this lesson.
Storing data
The raw data you get back from the sequencing center is the foundation of your sequencing analysis. You need to keep this data, so that you can always come back to it if there are any questions or you need to re-run an analysis, or try a new analysis approach.
Guidelines for storing data
- Store the data in a place that is accessible by you and other members of your lab. At a minimum, you and the head of your lab should have access.
- Store the data in a place that is redundantly backed up. It should be backed up in two locations that are in different physical areas.
- Leave the raw data raw. You will be working with this data, but you do not want to modify this stored copy of the original data. If you modify the data, you’ll never be able to access those original files. We will cover how to avoid accidentally changing files in a later lesson in this workshop (see File Permissions).
Some data storage solutions
If you have a local high performance computing center or data storage facility on your campus or with your organization, those are ideal locations. Get in touch with the people who support those facilities to ask for information.
If you do not have access to these resources, you can back up on hard drives. Have two backups, and keep the hard drives in different physical locations.
You can also use resources like Amazon S3, Microsoft Azure, Google Cloud or others for cloud storage. The open science framework is a free option for storing files up to 5 GB. See more in the lesson “Introduction to Cloud Computing for Genomics”.
Summary
Before analysis of data has begun, there are already many potential areas for errors and omissions. Keeping organized and keeping a critical eye can help catch mistakes.
One of Data Carpentry’s goals is to help you achieve competency in working with bioinformatics. This means that you can accomplish routine tasks, under normal conditions, in an acceptable amount of time. While an expert might be able to get to a solution on instinct alone - taking your time, using Google or another Internet search engine, and asking for help are all valid ways of solving your problems. As you complete the lessons you’ll be able to use all of those methods more efficiently.
Where to go from here?
More reading about core competencies
L. Welch, F. Lewitter, R. Schwartz, C. Brooksbank, P. Radivojac, B. Gaeta and M. Schneider, ‘Bioinformatics Curriculum Guidelines: Toward a Definition of Core Competencies’, PLoS Comput Biol, vol. 10, no. 3, p. e1003496, 2014.
- Data being sent to a sequencing center also needs to be structured so you can use it.
- Raw sequencing data should be kept raw somewhere, so you can always go back to the original files.
Content from Examining Data on the NCBI SRA Database
Last updated on 2023-11-20 | Edit this page
Estimated time: 30 minutes
Overview
Questions
- How do I access public sequencing data?
Objectives
- Be aware that public genomic data is available.
- Understand how to access and download this data.
In our experiments we usually think about generating our own sequencing data. However, almost all analyses use reference data, and you may want to use it to compare your results or annotate your data with publicly available data. You may also want to do a full project or set of analyses using publicly available data. This data is a great, and essential, resource for genomic data analysis.
When you come to publish a paper including your sequencing data, most journals and funders require that you place your data on a public repository. Sharing your data makes it more likely that your work will be re-used and cited. It helps to prepare for this early!
There are many repositories for public data. Some model organisms or fields have specific databases, and there are ones for particular types of data. Two of the most comprehensive public repositories are provided by the National Center for Biotechnology Information (NCBI) and the European Bioinformatics Institute (EMBL-EBI). The NCBI’s Sequence Read Archive (SRA) is the database we will be using for this lesson, but the EMBL-EBI’s Nucleotide Archive (ENA) is also useful. The general processes are similar for any database.
Accessing the original archived data
The sequencing dataset (from Tenaillon, et al. 2016) adapted for this lesson was obtained from the NCBI Sequence Read Archive, which is a large (~27 petabasepairs/2.7 x 10^16 basepairs as of April 2019) repository for next-generation sequence data. Like many NCBI databases, it is complex and mastering its use is greater than the scope of this lesson. Very often there will be a direct link (perhaps in the supplemental information) to where the SRA dataset can be found. We are only using a small part of these data, so a direct link cannot be found. If you have time, go through the following detailed description of finding the data we are using today (otherwise skip to the next section).
Locate the Run Selector Table for the Lenski Dataset on the SRA
See the figures below for how information about data access is provided within the original paper.

The above image shows the title of the study, as well as the authors.
The excerpt from the paper below includes information on how to locate the sequence data. In this case, the text appears just before the reference section.
Author Information All sequencing data sets are available in the NCBI BioProject database under accession number PRJNA294072. The breseq analysis pipeline is available at GitHub (http://github.com/barricklab/breseq). Other analysis scripts are available at the Dryad Digital Repository (http://dx.doi.org/10.5061/dryad.6226d). R.E.L. will make strains available to qualified recipients, subject to a material transfer agreement. Reprints and permissions information is available at www.nature.com/reprints. The authors declare no competing financial interests. Readers are welcome to comment on the online version of the paper. Correspondence and requests for materials should be addressed to R.E.L. (lenski at msu.edu)
At the beginning of this workshop we gave you experimental information about these data. This lesson uses a subset of SRA files, from a small subproject of the BioProject database “PRJNA294072”. To find these data you can follow the instructions below:
Notice that the paper references “PRJNA294072” as a “BioProject” at NCBI. If you go to the NCBI website and search for “PRJNA294072” you will be shown a link to the “Long-Term Evolution Experiment with E. coli” BioProject. Here is the link to that database: https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA294072.
Once on the BioProject page, scroll down to the table under “This project encompasses the following 15 sub-projects:”.
In this table, select subproject “PRJNA295606 SRA or Trace Escherichia coli B str. REL606 E. coli genome evolution over 50,000 generations (The University of Texas at…)”.
This will take you to a page with the subproject description, and a table “Project Data” that has a link to the 224 SRA files for this subproject.
Click on the number “224” next to “SRA Experiments” and it will take you to the SRA page for this subproject.
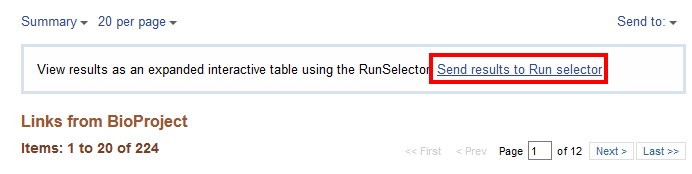
For a more organized table, select “Send results to Run selector”. This takes you to the Run Selector page for BioProject PRJNA295606 (the BioProject number for the experiment SRP064605) that is used in the next section.
Download the Lenski SRA data from the SRA Run Selector Table
Make sure you access the Tenaillon dataset from the provided link: https://trace.ncbi.nlm.nih.gov/Traces/study/?acc=SRP064605. This is NCBI’s cloud-based SRA interface. You will be presented with a page for the overall SRA accession SRP064605 - this is a collection of all the experimental data.
Notice on this page there are three sections. “Common Fields” “Select”, and “Found 312 Items”. Within “Found 312 Items”, click on the first Run Number (Column “Run” Row “1”).
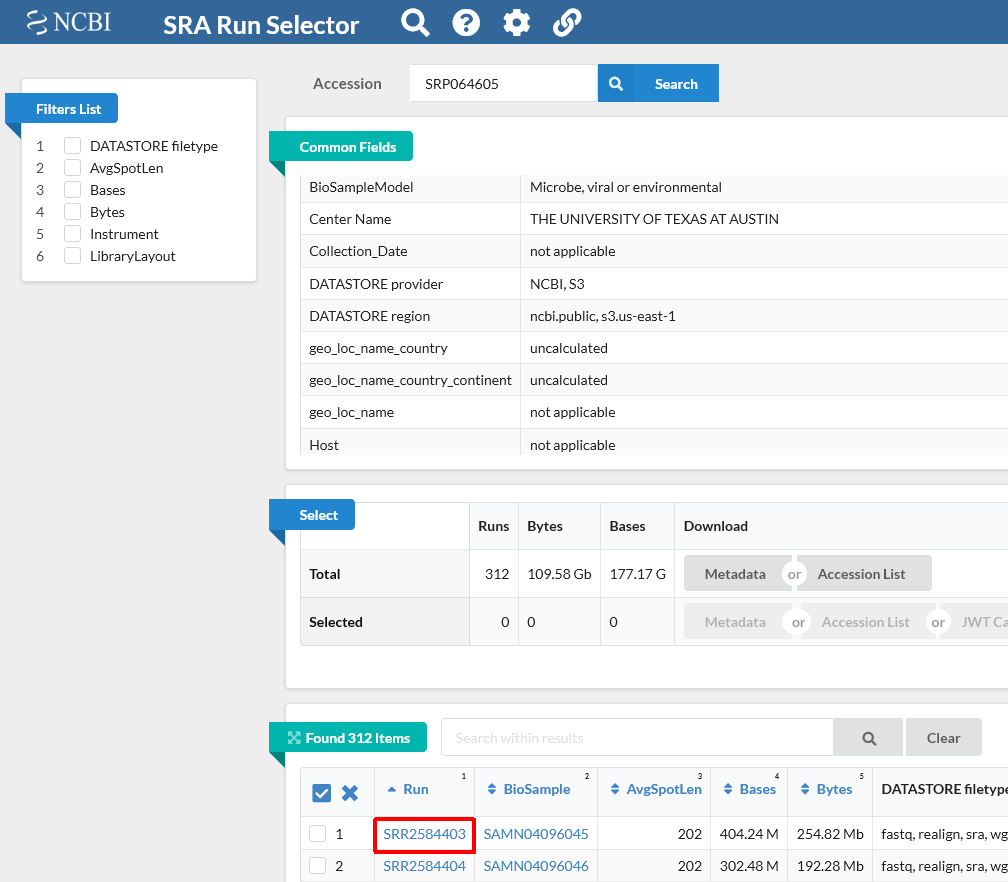
This will take you to a page that is a run browser. Take a few minutes to examine some of the descriptions on the page.
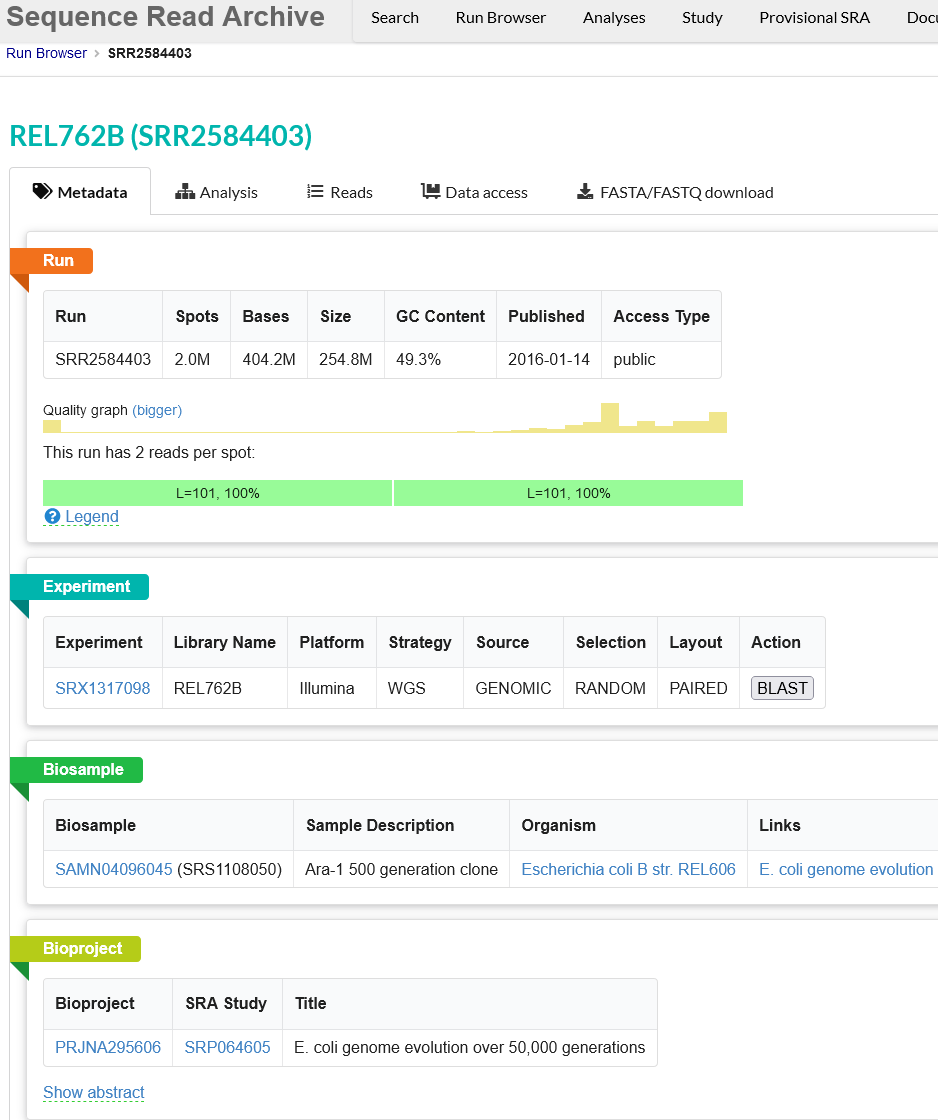
Use the browser’s back button to go back to the ‘previous page’. As shown in the figure below, the second section of the page (“Select”) has the Total row showing you the current number of “Runs”, “Bytes”, and “Bases” in the dataset to date. On 2022-12-06 there were 312 runs, 109.58 Gb data, and 177.17 Gbases of data.
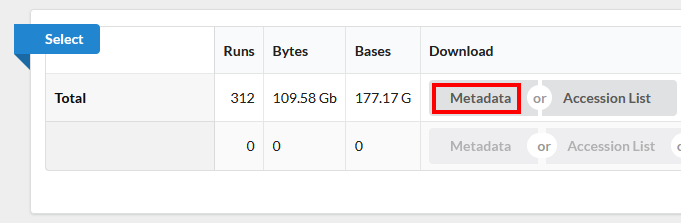
Click on the “Metadata” button to download the data for this lesson. The filename is “SraRunTable.txt” and save it on your computer Desktop. This text-based file is actually a “comma-delimited” file, so you should rename the file to “SraRunTable.csv” for your spreadsheet software to open it correctly.
You should now have a file called
SraRunTable.csv on your desktop.
Now you know that comma-separated (and tab-separated) files can be designated as “text” (
.txt) files but use either commas (or tabs) as delimiters, respectively. Sometimes you might need to use a text-editor (e.g. Notepad) to determine if a file suffixed with.txtis actually comma-delimited or tab-delimited.
Review the SraRunTable metadata in a spreadsheet program
Using your choice of spreadsheet program, open the
SraRunTable.csv file.
Discussion
Discuss with the person next to you:
- What strain of E. coli was used in this experiment?
- What was the sequencing platform used for this experiment?
- What samples in the experiment contain paired end sequencing data?
- What other kind of data is available?
- Why are you collecting this kind of information about your sequencing runs?
- Escherichia coli B str. REL606 shown under the “organism” column. This is a tricky question because the column labeled “strain” actually has sample names
- The Illumina sequencing platform was used shown in the column “Platform”. But notice they used multiple instrument types listed under “Instrument”
- Sort by LibraryLayout and the column “DATASTORE_filetype” shows that “realign,sra,wgmlst_sig” were used for paired-end data, while “fastq,sra” were used for all single-end reads. (Also notice the Illumina Genome Analyzer IIx was never used for paired-end sequencing)
- There are several columns including: megabases of sequence per sample, Assay type, BioSample Model, and more.
- These are examples of “metadata” that you should collect for sequencing projects that are sent to public databases.
After answering the questions, you should avoid saving any changes
you might have made to the metadata file. We do not want to make any
changes. If you were to save this file, make sure you save it as a
text-based .csv file format.
Downloading a few sequencing files: EMBL-EBI
The SRA does not support direct download of fastq files from its webpage. However, the European Nucleotide Archive does. Let’s see how we can get a download link to a file we are interested in.
Navigate to the ENA.
Near the top right, in the box next to “View”, type in
SRR2589044and click the “View” button.This will take you to a page with information about the data. Near the bottom you will have the option to download the data by FTP. You could download the
.fastqread files here, but we do not need to download these files right now and they are large. Alternatively, right click and copy the URL to save it for later.
We do not recommend downloading large numbers of sequencing files
this way. For that, the NCBI has made a software package called the
sra-toolkit. However, for a couple files, it’s often easier
to go through the ENA.
Where to learn more
About the Sequence Read Archive
- You can learn more about the SRA by reading the SRA Documentation
- The best way to transfer a large SRA dataset is by using the SRA Toolkit
References
Tenaillon O, Barrick JE, Ribeck N, Deatherage DE, Blanchard JL, Dasgupta A, Wu GC, Wielgoss S, Cruveiller S, Médigue C, Schneider D, Lenski RE. Tempo and mode of genome evolution in a 50,000-generation experiment (2016) Nature. 536(7615): 165–170. Paper, Supplemental materials Data on NCBI SRA: https://trace.ncbi.nlm.nih.gov/Traces/sra/?study=SRP064605 Data on EMBL-EBI ENA: https://www.ebi.ac.uk/ena/data/view/PRJNA295606
- Public data repositories are a great source of genomic data.
- You are likely to put your own data on a public repository.